


Indirect ELISA – This is a general protocol in which antigen coating and blocking may not be required if the wells from the manufacturer have been pre-adsorbed with the antigen.
1. Antigen Coating
- Dilute purified antigens to a final concentration of 1-10 μg/mL in bicarbonate/carbonate antigen-coating buffer (100 mM NaHCO3 in deionized water; pH adjusted to 9.6).
- Pipette 100 μL of diluted antigen to each well of a microtiter plate.
- Cover the plate with adhesive plastic and incubate at 4°C overnight (or 37°C for 30 min).
- Remove the coating solution and wash the plate 3X with 200 μL PBS (Phosphate Buffered Saline) buffer (10 mM Na2HPO4 and 1.8 mM NaH2PO4 in deionized water with 0.2% Tween 20; pH Adjusted to 7.4) with for 5 minutes each time. The coating/washing solutions can be removed by flicking the plate over a sink. The remaining drops can be removed by patting the plate on a paper towel or by aspiration. Do not allow the wells to dry out at any time.
2. Blocking
- Pipette 200 μL blocking buffer (5% w/v non-fat dry milk in PBS buffer) per well to block residual protein-binding sites. Alternatively, BSA or BlockACE can be used to replace non-fat dry milk.
- Cover the plate with adhesive plastic and incubate for 1-2 hour(s) at 37°C (or at 4°C overnight).
- Remove the blocking solution and wash the plate 2X with 200 μL PBS for 5 minutes each time. Flick the plate and pat the plate as described in the coating step.
3. Reagent Preparation
Prepare for the diluted standard solutions as shown on p.9.
4. Primary Antibody Incubation
- Serially dilute the primary antibody of choice with blocking buffer. The optimal dilution should be determined by a titration assay according to the antibody manufacturer.
- Pipette 100 μL of each diluted antibody per well. Repeat in duplicate or triplicate for accuracy. The negative control should be species- and isotype-matched as well as non-specific immunoglobulin diluted in PBS buffer.
- Cover the plate with adhesive plastic and incubate for 1 hour at 37°C (or 2 hours at room temperature). These incubation times should be sufficient to receive a strong signal. However, if a weak signal is observed, perform incubation overnight at 4°C for a stronger signal.
- Remove the diluted antibody solution and wash the wells 3X with 200 μL PBS for 5 min each time. Flick the plate and pat the plate as described in the coating step.
Read Now: ELISA SANDWICH?
5. Secondary Antibody Incubation
- Serially dilute the conjugated secondary antibody with blocking buffer immediately before use. The optimal dilution should be determined by a titration assay according to the antibody manufacturer.
- Pipette 100 μL of diluted secondary antibody solution to each well.
- Cover the plate with adhesive plastic and incubate for 2 hours at room temperature.
- Remove the content in the wells and wash them 3X with 200 μL PBS buffer for 5 min each time. Flick the plate and pat the plate as described in the coating step.
6. Substrate Preparation
Prepare the substrate solution immediately before use or bring the pre-made substrate to room temperature. The two widely used enzymes for signal detection are horse radish peroxidase (HRP) and alkaline phosphatase (AP), and their corresponding substrates, stopping solutions, detection absorbance wavelengths and color developed are as follows:
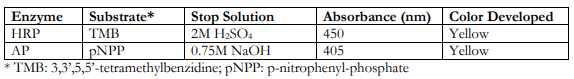
Note (Indirect ELISA):
- The TMB substrate must be kept at 37°C for 30 min before use.
- Hydrogen peroxide can also act as a substrate for HRP.
- Sodium azide is an inhibitor of HRP. Do not include the azide in buffers or wash solutions if HRP-labeled conjugate is used for detection.
7. Signal Detection
- Pipette 90 μL of substrate solution to the wells with the control and standard solutions.
- Incubate the plate at 37°C in the dark. If TMB is used, shades of blue will be observed in the wells with the most concentrated solutions. Other wells may show no obvious color.
- Color should be developed in positive wells after 15 min. After sufficient color development, pipette 100 μL of stop solution to the wells (if necessary).
- Read the absorbance (OD: Optical Density) of each well with a plate reader.
8. Data Analysis
- Prepare a standard curve using the data produced from the diluted standard solutions. Use absorbance on the Y-axis (linear) and concentration on the X-axis (log scale).
- Interpret the sample concentration from the standard curve.
Handbook Indirect ELISA by Bosterbio






